
Working from home, sometimes in pajamas, Ada Sedova taps into the world’s most powerful supercomputer in the hunt for a tiny molecule that could stop the coronavirus from infecting someone with COVID-19.
“I’m getting more done than ever, and with all the anxiety around the pandemic, I’m devoting a lot of my personal time to this effort,” said Sedova, a biophysics researcher at the Oak Ridge National Laboratory.
Her efforts could bring a 10-figure payday — specifically 2 billion molecular tests executed in just 24 hours.
Sedova seeks a ligand, an organic molecule less than a few dozen atoms in size. The right ligand will attach itself to a protein from the coronavirus, preventing it from infecting healthy cells.
The problem is there are so many ligands and proteins to check, and they keep changing shapes as their atomic forces shift. It’s one heck of a tiny needle in a ginormous stack of billions of possible compounds.
It could take many years for experts in wet labs to try each of the possibilities. Even simulating them all on the 9,216 CPUs on Summit, ORNL’s supercomputer, could take four years. So Sedova and colleagues turned to Summit’s 27,648 NVIDIA GPUs to accelerate their efforts.
They started using the OpenCL version of AutoDock, an open source program for simulating how proteins and ligands bind that was developed at Scripps Research in collaboration with TU Darmstadt. The OpenCL version on GPUs improved the processing speed by 50x compared to CPUs.
CUDA Cuts to the Chase
With help from NVIDIA and Scripps Research, the team ported the code to CUDA so it could run on Summit, delivering an added benefit of another 2.8x speedup. Another researcher, Aaron Scheinberg of Jubilee Development, accelerated the work another 3x when he found a way to use OpenMP to speed up feeding data to the GPUs.
In another test, Sedova showed results that suggest they may be able to screen a dataset of 1.4 billion compounds against a protein with high accuracy in as little as 12 hours. That’s more than a 33x speedup compared to a program running on CPUs.
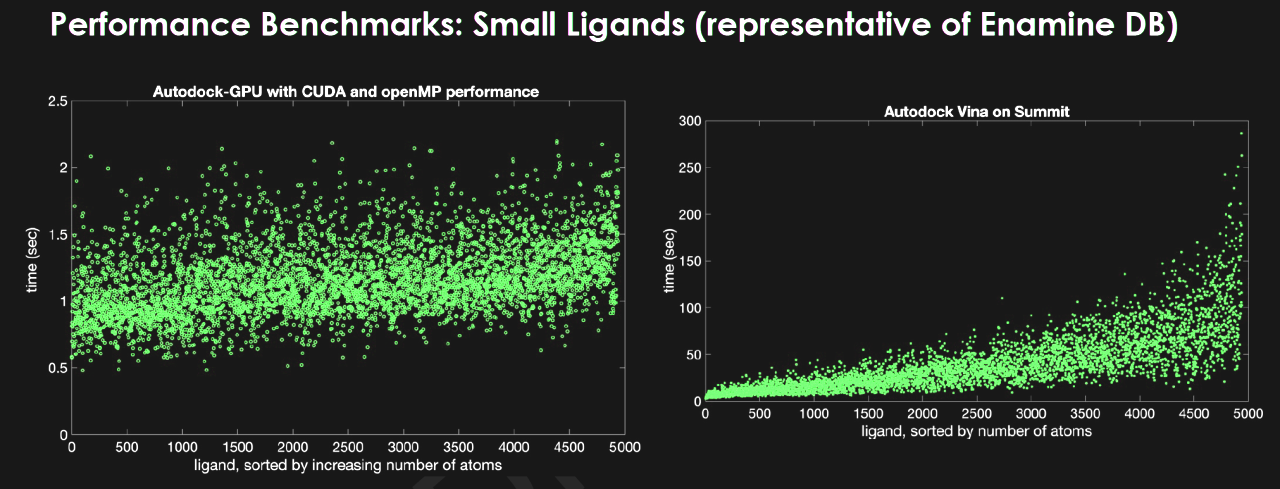
“GPUs combined with Summit’s scale and architecture provide the capability for docking billions more compounds than what was possible previously,” she said.
Another member of the team, biophysicist Josh Vermaas, gave a shout-out to NVIDIA’s Scott Le Grand, who helped port AutoDock to CUDA. “He’s been a phenomenal help in improving performance from what used to be an OpenCL-only code,” said Vermaas in a blog on the origins of the work.
Simulating 2 Billion Compounds in 24 Hours
Sedova now believes with further improvements the team could create a capability to examine as many as 2 billion compounds in 24 hours. It would mark the first simulation of that size at high resolution.
Researchers still face a few challenges getting to that milestone.
The standard workflow for protein-ligand docking uses a sluggish file-based process. It’s fine for tests of a few hundred compounds on a laptop, but at the scale of hundreds of thousands of files it could bog down even the world’s largest supercomputer.
That’s a call to action for open source developers who want to help accelerate science.
Sedova’s team is leading the charge, assembling a new workflow that promises to securely launch vast numbers of jobs on Summit. She’s consulting with the system’s I/O experts and trying to spin up a database to hold all the ligands.
The next step is launching an experiment with about 1 million compounds on 108 of Summit’s 4,608 nodes. “If it works, we’ll launch the big run with 1.4 billion compounds using all of Summit’s nodes,” she said.
Narrowing the Search for Promising Molecules
If the team succeeds, they’ll send researchers in Memphis a list of about 9,000 of the most promising compounds to test in their wet lab with the real virus. It’s not the needle in the haystack, but it’s a needle in a shovel of hay.
The work got its start in January when a top ORNL researcher, Jeremy C. Smith, showed the first work using the Summit supercomputer for drug research to fight the coronavirus. The work is still in its early days.
Looking ahead, Sedova has ideas for other ways to bridge the field of protein-ligand binding into the typical methods used in high performance computing. And she has plenty of energy for pursuing them, too.
Learn about more efforts using GPUs to fight the coronavirus on NVIDIA’s COVID-19 page.